[데일리메디 양보혜 기자] 내년부터 제네릭의약품 동등성, 제조방법 등에 대한 관리 강화를 통해 의약품 품질 신뢰도가 제고되면서 안전성·유효성 평가가 기존보다 훨씬 엄격해진다.
18일 식약처는 온라인 '2021년 의약품 허가정책 주요 개선 과제'를 발표했다. 주요 내용은 국내 허가 기준을 국제 조화를 이루기 위해 높인다는 것이다.
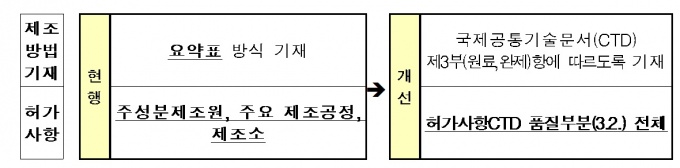
우선 전문의약품 제조방법을 국제공통기술문서(CTD)를 활용해 상세히 기술하고, 제조방법에 대한 관리도 강화된다.
CTD는 의약품 허가에 필요한 제출 자료를 국제적으로 표준화한 문서로서 의약품에 대한 일반적 설명, 품질, 비임상·임상시험 자료 등이 포함된다.
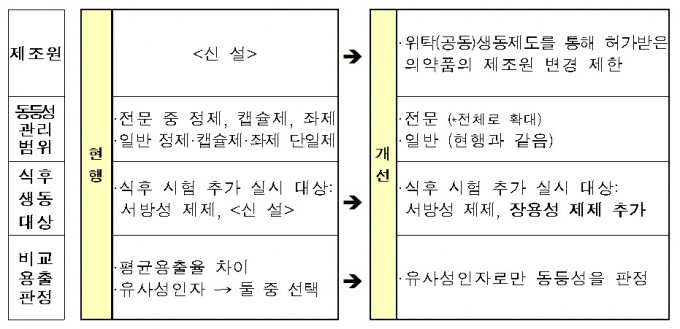
제네릭의약품 가운데 동등성 관리가 필요한 품목을 일부 제형에서 전체 제형으로 확대한다. 전문의약품의 경우 기존에는 정제, 캡슐제, 좌제 등에 적용됐지만, 내년부터는 전문의약품 전체에 포함된다.
올해 10월부터 개정이 돼 경구용 제제 전문 의약품은 1년 6개월, 무균제제는 2년 이후 전문의약품 전체는 3년 이후 적용이다.
김영주 의약정책과 서기관은 "새롭게 포함되는 장용성제제의 경우 식후 생동성시험 평가를 위한 제출 요건으로 추가하고, 비교용출 시험결과의 판정 방법을 유사성인자(f2)로 한정하는 등 국제 규제와 조화를 통해 국내 제네릭의약품의 품질 경쟁력을 강화할 예정"이라고 설명했다.
전문의약품 중 복합제도 제품명에 3개 이하 유효성분을 함께 표시해야 한다. 기허가 제품에 대한 소급적용은 하지 않고, 새로 허가 받는 제품에 대해서만 적용된다.
김영주 서기관은 "주성분은 명칭이 너무 길어서 염이 붙은 부분은 제외하고, 유효성분만 표시해주면 된다"며 "새로 허가 받은 제품에만 변경사항이 적용된다"고 말했다.
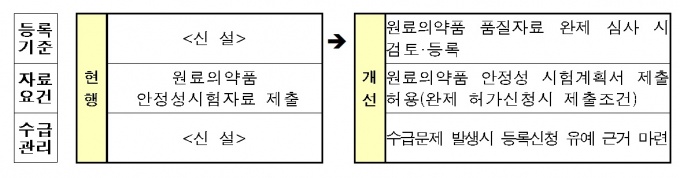
설계기반 품질 고도화(QbD) 제도를 도입해 제품 출하 시 타당성을 입증할 경우 일부 시험을 생략할 수 있도록 하고, 완제의약품과 원료의약품 간 연계 심사도 진행하다.
완제의약품 허가신청 시 제출하는 것을 조건으로 원료의약품에 대한 안전성시험 계획서 제출을 허용하는 등 규제를 합리적으로 개선한다.
의약품 안전성·유효성 평가도 까다로워진다. 특히 외국 의약품집에 수재된 경우에도 자료 제출을 의무화해 과학적 자료에 기반한 안전성·유효성 평가체계를 구축한다.
현행 규정에 따르면 전문의약품은 3년이내 발간된 외국 의약품집 수재 시 독성,약리자료 면제되지만, 이 조항이 삭제돼 향후에는 해당 자료를 제출해야 한다.
일회용 점안제의 안전사용을 위해 1회 사용 적정용량(0.5밀리리터 이하) 제한 기준도 설정했다.
세계보건기구(WHO)의 국제 의약품 분류 코드를 품목허가 시 기재하는 등 정보 제공을 확대해 소비자 알권리도 강화한다.
김영주 의약정책과 서기관은 "이번 개선안에 대해 연내 행정예고를 할 예정이며, 바이오의약품은 해당되지 않는다"면서 "바뀌는 규정에 대한 업계 의견을 충분히 수렴하겠다"고 말했다.








 양보혜 기자 (
양보혜 기자 (
